阅读报告:The divergent roles of autophagy in ischemia and preconditioning
Compared & mixed with privious review Death and survival of neuronal and astrocytic cells in ischemic brain injury : a role of autophagy[2011]
Abstract
细胞自噬是一种高度规律化的生命进程,存在于酵母菌到哺乳动物等一系列生物中,其进程常常为通过溶酶体系统消化细胞内的细胞质小分子和细胞器,Short-life proteins are usually degraded via the ubiquitin-proteasome pathway, whereas the longlife proteins and organelles are primarily degraded via the autophagy-lysosome pathway .它经常发生在细胞饥饿、分化、正常生长的过程中,以维持内环境的稳定以保证细胞的生存。累计的证据表明,自噬参与了动物缺血、缺氧(HI/global and focal ischemia)下的脑损伤中的神经细胞死亡,目前。the contribution of autophagy to cell death/survival during these processes is still debated.
HI是自发凝结一种强大的刺激导致再灌注减少,在这种情况下,对于脑细胞的生存找成极大的挑战。过去的观点认为,HI损伤的范围取决于大脑的成熟程度、损伤的持续时间和严重性,认为新生的大脑比成年大脑更能够忍受OGD/HI环境,但是,临床数据和实验结果推翻了这一观点。
IPC(ischemic preconditioning,缺血预处理)是指组织、器官经受一次或者数次短暂的缺血再灌注后,能够提高对随后较长时间内IR损伤耐受能力的现象,被广泛用于动物缺血模型的研究上。普遍认为其与pkc和mapks的信号通路激活有关。[1]
本综述将列举此前的电镜、生物组化、蛋白质印迹(western blot)等证据、解释细胞自噬在神经细胞死亡中的角色,及其激活和作用机制,我们将探讨自吞噬、坏死、凋亡之间的关联,并揭示受损神经细胞的形态学,通过研究APL机制,希望能够进一步揭示其作用原理,为临床治疗中风等大脑缺氧缺血/再灌注疾病找到以APL为靶向的有效新疗法。
Different types of neural cell death in ischemic
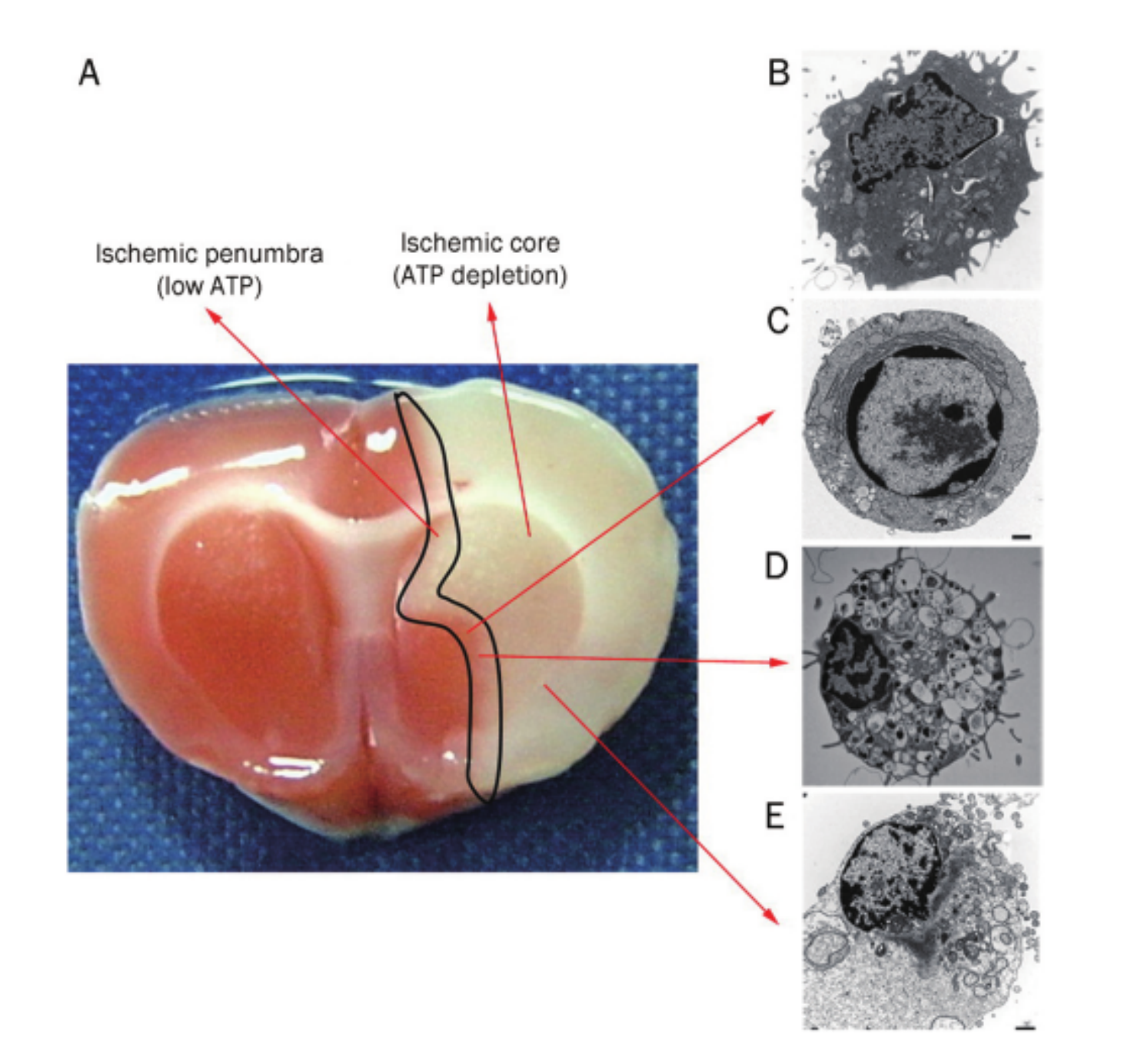
Necrosis, apoptosis and autophagy in focal cerebral ischemia.图B:正常细胞。
TTC染色表明ischemic core region 和 ischemic penumbra这两个区域(图A),图B -E 都分别是电镜图片。图片显示,坏死优越地表现在core region,而细胞自吞噬和凋亡则经常观察到出现在penumbra区域上在局部大脑缺血后。通过双免疫标记技术也观察到自噬标志物主要出现在损伤的边缘区域,但是也观察到标志物出现在损伤内的一些神经元细胞内。
坏死主要的形态学特征是细胞质液泡化和原生质膜降解(图E),坏死过去被认为是由于意外事件使得细胞能量消耗到不适合维持细胞生存的地步后,细胞被动的死亡,但是研究认为,坏死是一种细胞信号启动的生物进程。
凋亡的形态学特征是细胞核和细胞质皱缩,细胞破碎形成凋亡小体(图C),在生化层次上,凋亡是由于phosphatidy暴露和主要效应物caspase激活的,其途径包括线粒体途径和细胞外信号介导途径。研究已经证明,大脑缺血诱导的细胞延时死亡就是由凋亡进行的。
自噬的形态学特征表现为:空泡化、细胞质降解、轻微的染色质浓缩,在这一过程中,高尔基复合体、多核糖体、内质网的消化要优先于细胞核。在这个过程中,需要完整的线粒体提供能量、同时,完整的细胞骨架也是必须的,在分子层次上面, Atg7 is a critical upstream gene involved in multiple neu-ronal death pathways(induced caspase-3 activation and neuronal death).
Autophagy is activated in ischemic and/or hypoxic damaged brain tissue
研究人员在使用电镜分析一个沙鼠大脑缺血模型时观察到如下的情况,

3d后cathepsin-B-positive 溶酶体增高的同时,自噬小泡状的结构也在这一阶段同时增加,这说明这些增多溶酶体大部分都是自噬小泡。
2006年,在一侧颈总动脉堵塞模型中,Adhami et al 指出,在组织水平上,HI导致永久的再灌注减少(血循环内血小板沉积和纤维蛋白积累);在细胞水平上,caspase-3并没有激活,并且很少细胞完成了凋亡过程,相反,许多损伤表现出autophagic/lysosomal cell death的特征。而在 2008, Carloni et al发现autophagy marker, Beclin1, 在HI发生后一小段时间内增加,在最近,他发现皮质、海马体内HI的发展不仅是由于LC3-II增加导致的自噬泡增多,同时也有溶酶体活动(cathepsin D, acid phosphatase, and β-N-acetylhexosaminidase)的参与。western blot 分析同样发现在缺血大脑皮质中LC3-II和cathepisn B(一种大脑实质内溶酶体的主要蛋白酶)的增加。
take togethor
Autophagy was activated in both the ischemia and reperfusion phases during cerebral I-R. Autophagy plays a detrimental role in permanent ischemia, whereas autophagy during reperfusion may contribute to neuroprotection. Inhibition of autophagy in the reperfusion phase aggravate brain injury induced by I-R .
autophagy in neurons
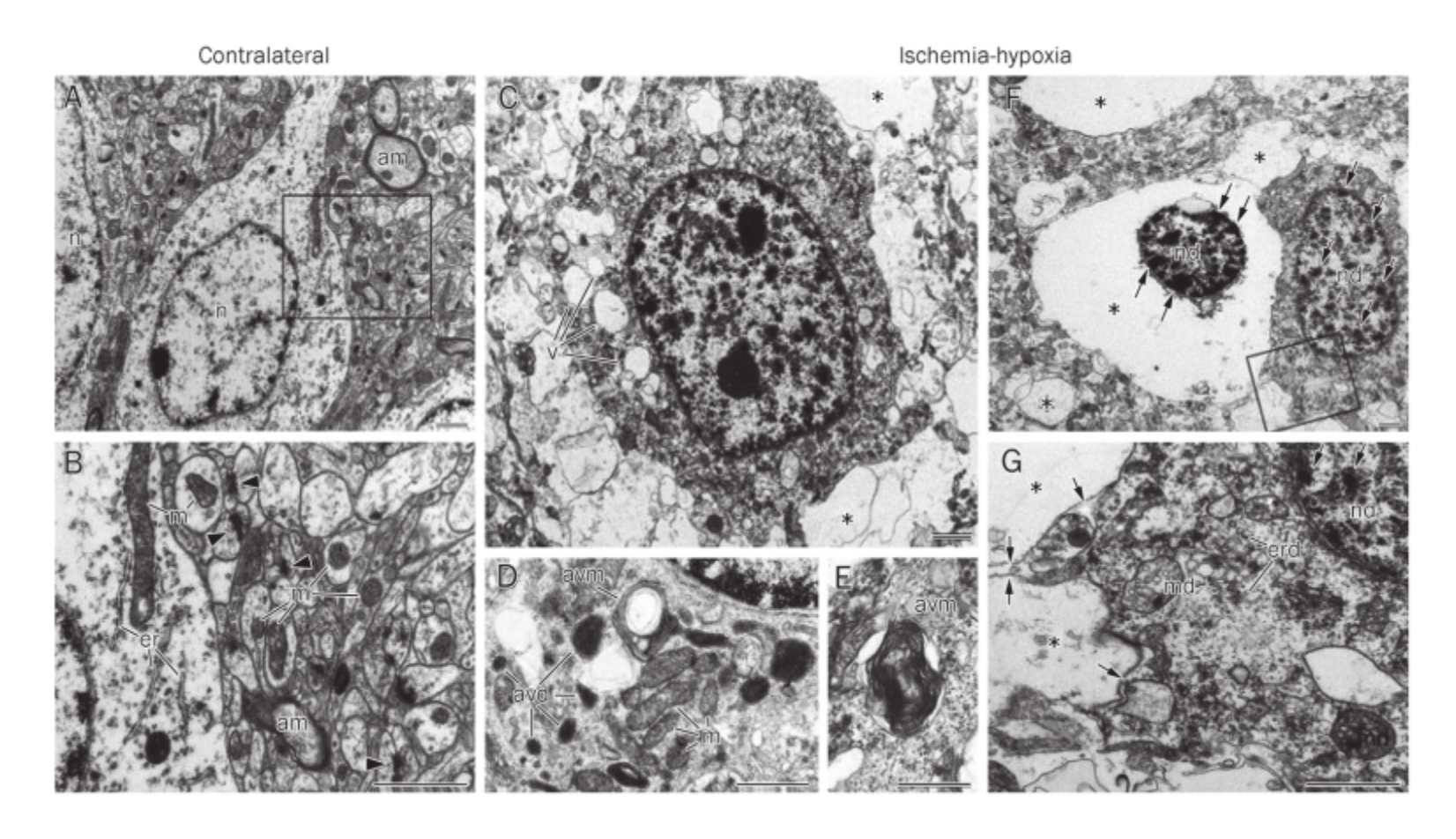
in an adult mouse HI model, the activation of autophagy in different degrees of neuronal injury has been examined in detail. Cortical neurons on the challenged side show vacuole-associated damage rang-ing from cells harboring multiple cytoplasmic vacuoles (Figure2C) to cells completely lacking cytoplasmic contents (Figure2F). There is also diffuse myelin degeneration and loss of syn-apses (Figure 2, compare B with G). In less damaged neurons,judging from the morphology of healthy mitochondria, there are many vacuole-related structures containing electron-dense material (Figure 2D, avd) or whorls of membranous material (Figure 2D and 2E; avm).More severely damaged neurons show condensed chromatin in the nucleus, cup-shaped endo-plasmic reticulum fragments, swollen mitochondria, and vacuoles that contain little material (Figure 2G, asterisk) in the cytoplasm. Moreover, many severely damaged neurons exhibit a shrunken nucleus with condensed chromatin sur-rounded by an extensively lysed cytoplasm but retain an intact plasma membrane (Figure 2F).
这些形态学特征提示这是自噬诱导的细胞死亡程序,在这个过程中,1h:自噬泡出现;3-12h:自噬泡增加,吞噬了细胞器的囊泡可以观察到。
autophagy in astrocytes
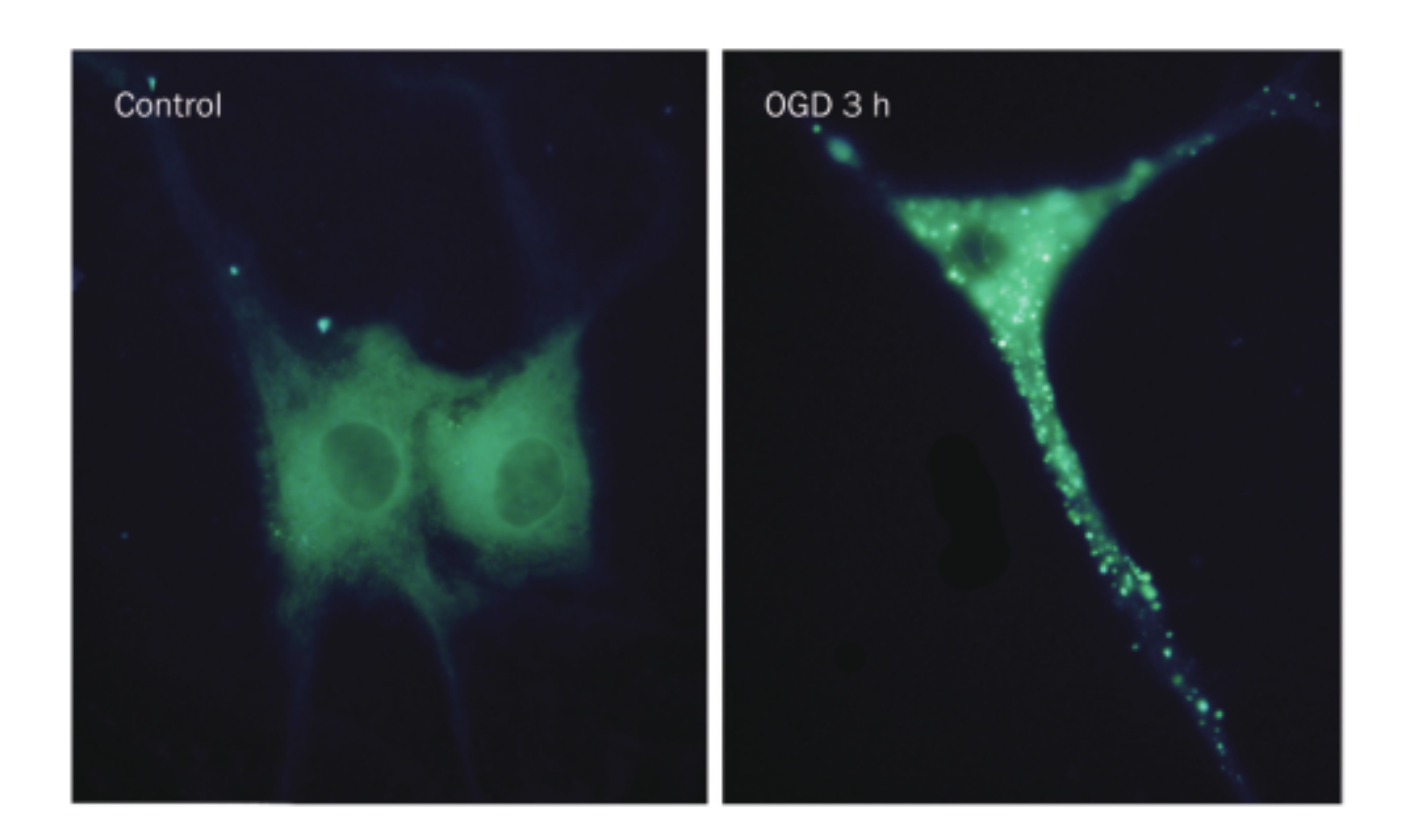
Increased autophagic vacuoles in primary astrocytes after OGD injury. Astrocytes were cultured and labeled with MDC.
荧光点在OGD后0.5h出现,并在3h时候达到顶峰。western bot检测到autophagy-related proteins LC3、Beclin1、lysosomal-related protein cathepsin B 、the lysosome associated membrane protein 2 (LAMP2)均增高。
Take together
The autophagy/lyso-some pathway (ALP) is activated in the ischemic astrocytes in both in vivo and in vitro ischemic or hypoxic models .
Mechanisms of autophagy activation in ischemic…
Ischemic brain injury causes death in both neurons and astrocytes .endoplasmic reticulum (ER) stress and the AMPK/TSC/mTOR, Beclin 1/BNIP3/SPK2 and FoxO/NF-κB transcription factors have been implicated as major injury mechanisms.
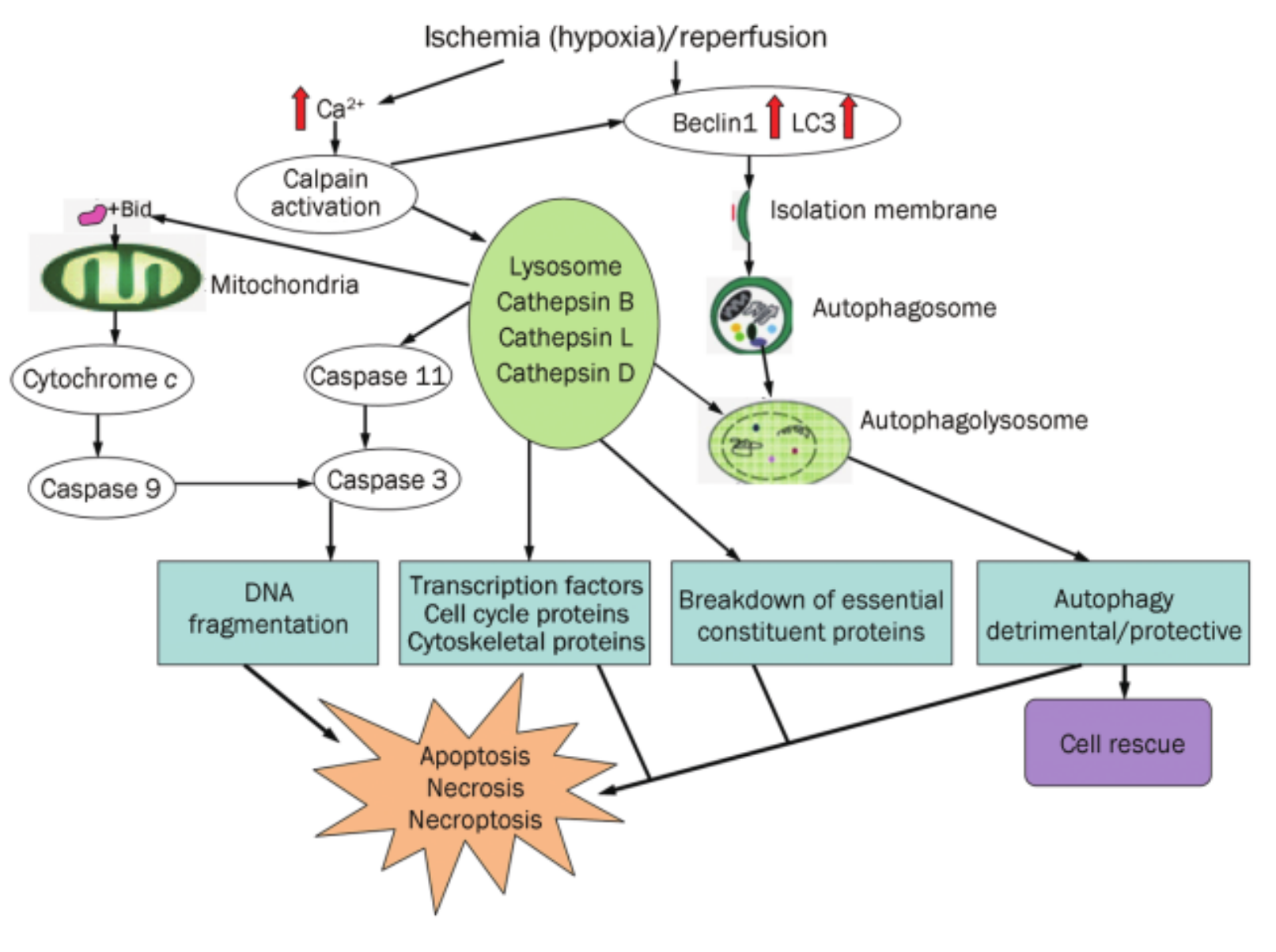
pathways,APL activation(VERSION 2011) either rescues the injured neural cells or induces cell death by necrosis, necroptosis, or apoptosis.Autophagy activation is often accompanied by increased lysosomal hydrolases (cathepsins),whereas cathepsins are involved in caspase activation and the subsequent apoptosis process. Cathepsin B activates caspase-3 via caspase-11 during cell apoptosis
ER stress:Hypoxia activates the ER-resident translation initiation factor eIF2α and PERK, and eIF2α kinaseregulates starvation- and virus-induced autophagy. ER stress, which includes the unfolded protein response (UPR) initiated by cell stress conditions . A major pathway of UPR is the suppression of most protein translations through the phosphorylation of eukaryotic translation initiation factor 2 subunit a (eIf2a) via the PKR-like ER kinase(PERK). Another pathway is the upregulation of the expression of ER-localized molecular chaperones, such as glucose-regulated protein 78(G RP78/Bip, a key mediator of autophagy during preconditioning ), GRP94, and other molecu-lar chaperons, such as heat shock proteins (HSPs). ER stress also induces ER associated protein degradation (ERAD) to monitor and eliminate unfolded/misfolded proteins in the ER. ERAD includes the ubiquitin/proteasome pathway (ERAD I) and the autophagy/lysosomal pathway (ERAD II). When ERAD I is injured,the autophagy/lysosomal pathway functions as ERAD II to degrade abnormal protein aggregates.prolonged ER stress may activate an ER stress-dependent apoptotic pathway by the induction of caspases-12 and CHOP (C/EBP) homologous protein,growth arrest and DNA damage inducible gene 153, GADD153).
Oxidative stress activates autophagy in dopaminergic(多巴胺型的) neuronal cell lines and cultured(培养的) primary astrocytes.mild ER stress promotes the recovery of ER functionand cell survival during subsequent lethal ischemia.
AMPK/TSC/mTOR-dependent APL:The AMP-activated protein kinase (AMPK) is an evolutional conserved eukaryotic protein kinase perceiving the cellular energy status. During energetic stress (an increased AMP/ATP ratio), AMPK is activated and phosphorylates TSC2, leading to the inhibition of mTOR (mammalian target of rapamycin), which plays a role in gating the processof autophagy .
mTOR is the catalytic subunit of two functional complexes:mTORC1 and mTORC2. Rictor (the rapamycin-insensitive companion of mTOR) is the component of mTORC2 responsible for the phosphorylation of Akt by binding to mTOR.Rapamycin specifically inhibits mTORC1, thus inducing autophagy in many cell types AND exerts neuroprotection against cerebral ischemia.resveratrol attenuates the activationof mTORC1, low-dose resveratrol was shown to significantlyinduce the expression of Rictor, a component of mTORC2,and activate its downstream survival kinase Akt (Ser473).
Beclin 1/BNIP3/SPK2/BAG1-dependent APL:
BH3-only proteins and pharmacological BH3 mimetics induce autophagy by competi- tively disrupting the interaction between Beclin 1 and Bcl-2 or Bcl-XL
An increase in Bcl-2 phosphorylation was associated with decreased interactions between Bcl-2 and Beclin 1 and an increased expression of LC3-II,BNIP3 (Bcl-2/adenovirus E1B interacting protein 3, 19 kDa) is a mitochondrial pro-cell death Bcl-2 family member with a BH3-domain.Hypoxia upregulates BNIP3 via HIFlα. The upregulated BNIP3 then displaces Beclin 1 from Bcl-2/Beclin 1 or Bcl-XL/Beclin 1 complexes, releasing Beclin 1 and thereby initiating mitochondrial autophagy and decreasing the production of reactive oxygen species.
Beclin 1, the mammalian homologue of the yeast Atg6, is an autophagy-associated tumor suppressor.
Sphingosine kinase 2 (SPK2) regulates the level of intracellular sphingosine 1-phosphate (S1P) and has been shown to play a role in preconditioning . SPK2 contributes to isoflurane and hypoxic preconditioning-induced autophagy activation both in vivo and in vitro.a possibility that SPK2 is another BH3-only protein that is upregulated by preconditioning and that can displace Beclin 1 from Bcl-2/Beclin 1 complexes, thus releasing Beclin 1 and initiating autophagy.
.Ischemic preconditioning activates autophagy and increases BAG-1 expression. BAG-1 modulates the chaperone activities of HSP70 and HSC70 by binding with HSP70/HSC70 and participates in the induction of autophagy .Co-immunoprecipitation and co-immunofluorescence analyses revealed that LC3-II binds with BAG-1.Moreover, another BAG family member, BAG-3,is responsible for the induction of macroautophagy in association with HSPB8. Thus, BAG family members are involved in the induction of autophagy for the degradation of damaged or oxidized proteins to promote cell survival.
kinase C-dependent:The induction of ER stress by thapsigargin or tunicamycin induced PKCθ phosphorylation and translocation to LC3-containing dotstructures, and then elicited autophagy,Ca 2+ -dependent PKCθ activation is specifically required for autophagy in response to ER stress
FoxO/NF-κB transcription factors:Nuclear factor kappa B (NF-κB) is released from the inhibitory protein IκB and then translocates to the nucleus to initiate gene expression involved in a broad range of biological processes.Deletion of the NF-κB p50 subunit increases the number of Beclin 1/TUNEL-positive neurons/vascular endothelial cells in the ischemic cortex. The increased autophagic cell death is accompanied by a downregulation of Akt-mTOR signaling.Increased FoxO1 activity preferentially activates Atg12 gene expression, whereas FoxO3 preferentially activates expression of the LC3 gene.FoxO may also induce the expression of BNIP3,which then displaces the autophagic effector Beclin 1 from inactive Bcl-XL complexes to initiate autophagy, and JNK may be a potent negative regulator of FoxO-dependent autophagy in neurons.
Glutamate(谷氨酸) excitotoxicity mediated by N-methyl-D-asparticacid (NMDA) receptor activation plays a key role in many aspects of cerebral ischemic injury. Excitotoxicity is the basis for necrotic cell death in the ischemic core and the initiator(启动者) of programmed cell death in the ischemic penumbra.NMDA agonists(受体) induce(诱导) the activation(激活) of autophagy in damaged neurons in the striatum . Excitotoxicity-mediated activation of autophagy may be associated with the activation of the c-JunN-terminal kinase (JNK) signaling pathway, which is one of the most important NMDA receptor-mediated signal pathways. in the jnk protein family, D-JNKI1 has also been found to have a potential neuroprotective effect in aclinically relevant model of neonatal cerebral HI and to reduce autophagosome formation in the thalamus, suggesting that the JNK signaling pathway involves HI-induced autophagic cell death.
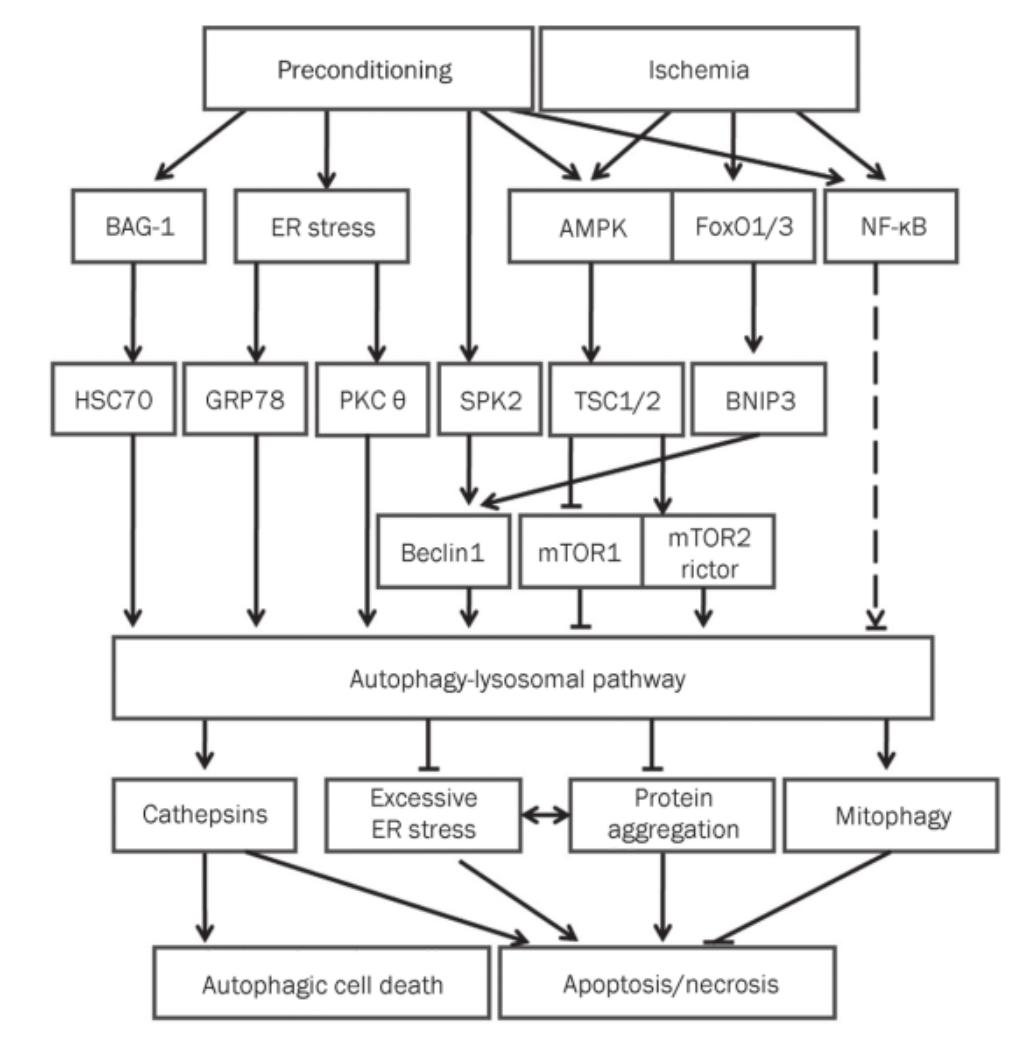
APL version 2015,These signaling pathways may act together in a coordinated manner rather than independently and form a complex network to regulate autophagy activation.
Contributions of autophagy activation to neural cell survival and death in ischemic and/or hypoxic cerebral injury
Excessive autophagy induces cell death via direct autophagic cell death or indirect crosstalk with apoptosis, whereas mild autophagy may remove damaged organelles or abnormal protein to promote cell survival.The dividing line between the dual roles of autophagy remains unclear.
-
Mild or moderate activation of autophagy promotes neural cell survival.
3-MA reduce Beclin1 expression and switch the mechanism of cell death modes from poptosis to necrosis. Conversely, rapamycin, an inducer of autophagy, augments Beclin1 expression, diminishes necrotic cell death, and decreases brain injury after neonatal HI.
autophagy is increased in neuronal cells after neona-tal HI and suggest that the activation of autophagic pathways represents a potential protective mechanism in the early stages of brain injury.Beclin1 may pos-sess a repair function via autophagy.
-
Overactivation of autophagy promotes neural cell death.
3-MA inhibits fusion(融合) between autophagosomes and lyso-somes.3-MA and Baf significantly reduce the death of astrocytes(星形胶质细胞) and play a protective role in astrocytic injury induced by cerebral isch-emia.These neuroprotective effects are associated with the inhibition of the up-regulation of LC3-II and cathepsin B in the rat brain.evidence on the genetic level show-ing that ribonucleic acid interference (RNAi)-mediated down-regulation of Beclin1 decreases infarct volume and inhibits histological injury and neurological deficits induced by focal(局部) cerebral ischemia supports the present conclusion.
which modes of neural cell death ALP activation is involved in.
enhanced autophagy in delayed neuronal death after H-I is differentially linked to apoptosis according to the brain regions.
-
Simultaneous activation of autophagic and apoptotic mechanisms can occur in the same dying neuron.neurons presenting strong autophagic features in the border of the lesion 24 h after HI are TdT-mediated dUTP-biotin nick end labeling (TUNEL)-negative and display moderate chro-matin condensation without being pyknotic, which suggests that autophagy precedes apoptosis and might even initiateapoptosis.Ischemia-induced APL activation may cause neuronal andastrocytic apoptotic cell death through the cathepsin-caspase signaling pathway.On the other hand, there is a crosstalk between autophagy and apoptosis in Bcl-2 levels.Bcl-2 can block caspase-independent cell death and mitochondrial degradation and the two processes involved in autophagy.ischemia-induced APL activation may cause neuronal and astrocytic apoptotic cell death via regulating Bcl-2.
-
It remains to be determined if autophagy is actu-ally a response to necroptotic cell death.
Some suggest that autophagy blockage may interrupt the activation of the apoptotic process and switch the mechanism of cell death from apoptosis to necrosis.However, evidences show that excessive autophagosome formation is induced early during necrotic cell death in C elegans neurons and that autophagy is required for neuronal necrotic cell death.Autophagy synergizes with lysosomal catabolic mechanisms to further facilitate(促进) neuronal necrotic cell death.however,this needs to be further comfirmed in the many other animal models.
Result
Autophagy is an essential process for maintaining cell function by removing protein aggregates and damaged organelles.Autophagy contributes to ischemic tolerance induced by reconditioning.Preconditioning increased the expression of the autophagy-related protein LC3 and Beclin 1 and promoted the formation of autophagosomes.
Autophagy plays dual roles in determin-ing cell fate, depending on specific cell types and stimuli. It seems that physiological levels of autophagy caused by mild or moderate cerebral ischemia and/or hypoxia appear to be protective via the production of energy to prevent cell necrosis by catabolism and inhibit cell apoptosis by removing damaged mitochondria. However, high levels of autophagy caused by severe cerebral ischemia and/or hypoxia (focal cerebral isch-emia or severe HI) may lead to self-digestion and eventual cell death.
More work is required to determine the requirements under which the activation of autophagy reflects a protective or harmful response in cerebral ischemia.
Reference
- pkc介导p44/42mapks信号转导通路在肝脏缺血预处理保护效应中作用的研究,王瑜,2002